

From Signal to Chromatin to Metabolism: A Step-By-Step, Falsifiable Route to In-Vivo Gene Re-Expression
Introduction
The ability to reactivate silenced genes in vivo without permanent DNA editing represents a paradigm shift in translational medicine. Multi-locus gene re-expression holds potential for treating refractory cancers, metabolic disorders, and epigenetically-driven diseases. Emerging evidence suggests that the interplay between signal transduction, chromatin accessibility, and metabolic cofactor availability can create a permissive environment for transcriptional reactivation.
This framework proposes a step-wise, falsifiable approach leveraging three interdependent modules:
- PI3K modulation to reduce oncogenic signaling while preserving DNA repair competence
- SIN3/HDAC relief to transiently open chromatin at target loci
- α-Ketoglutarate (AKG)-driven support to fuel demethylase-mediated epigenetic remodeling
By integrating molecular readouts with functional phenotypes, this design allows rigorous testing of multi-locus, multi-chromosomal gene re-expression in a controlled, reproducible manner.
Experimental Design (Overview)
Cohort & Ethics
- IRB/ethics approval required
- Inclusion/exclusion criteria: e.g., receptor-negative TNBC post-standard-of-care
- Pre-registration of primary and secondary endpoints
Module A — Signal (PI3K)
Goal: Reduce PI3K tone while maintaining mTORC1-linked DNA repair
Rationale: PI3K/AKT/mTOR mediates DNA damage response; full blockade risks impaired repair or compensatory resistance.
Approach: Selective PI3K inhibition with downstream S6 phosphorylation monitoring
References: PubMed Central | Cell | BioMed Central
Module B — Chromatin (SIN3/HDAC)
Goal: Increase chromatin accessibility at target loci (e.g., ESR1, PGR)
Assays: ATAC-seq, ChIP-seq, SIN3A/B occupancy profiling
References: PubMed Central
Module C — Metabolism/Epigenetics (AKG)
Goal: Elevate α-ketoglutarate to enhance TET/JmjC demethylase activity
Assays: Global 5-hmC/5-mC profiling, locus-specific bisulfite sequencing, histone mark profiling, metabolomics
References: BioMed Central | PubMed
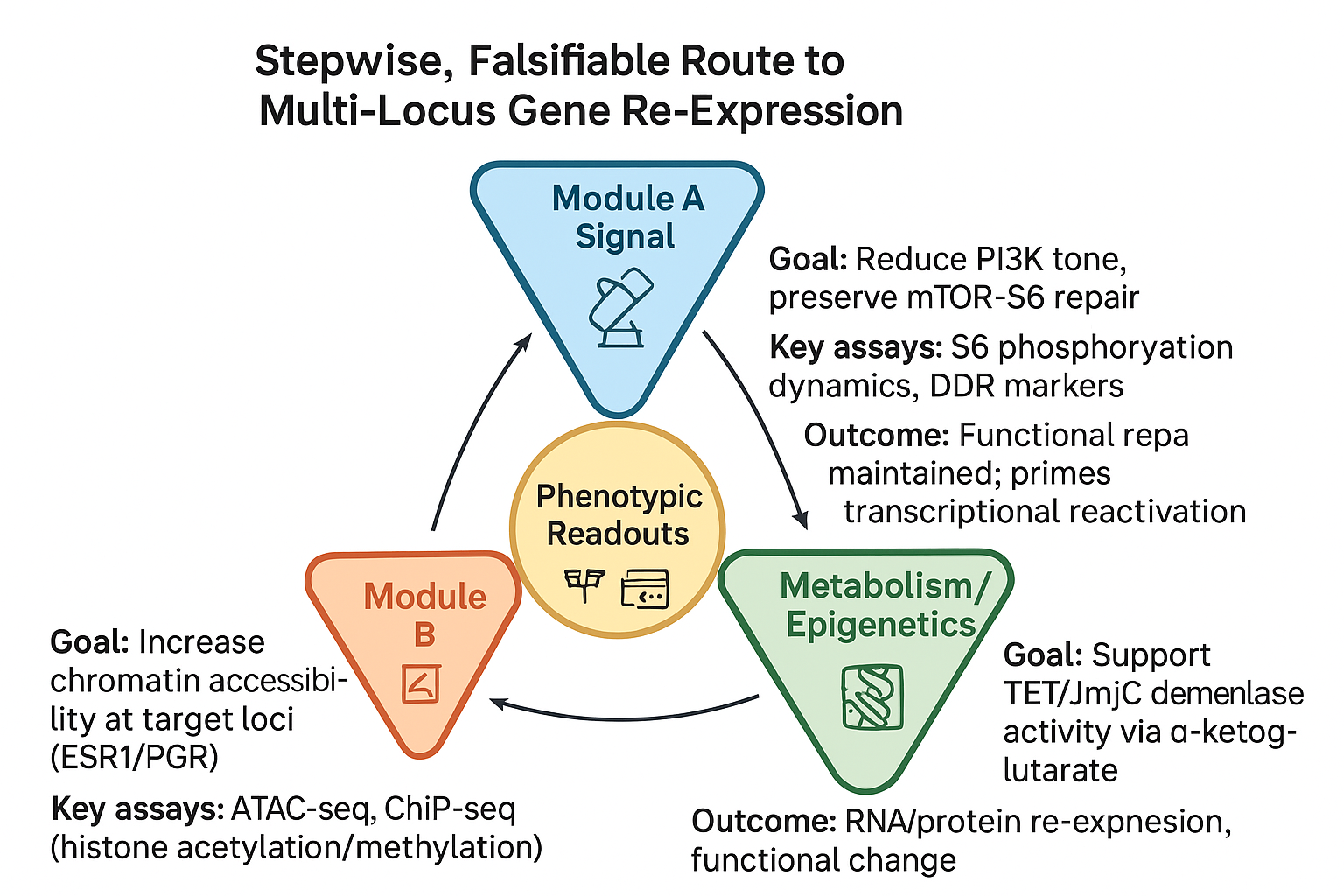
Integrated Readouts (Primary Endpoints)
- Transcriptional re-expression: qPCR / RNA-seq at silenced loci
- Protein re-expression: IHC for ESR1/PGR or other disease-relevant targets
- Functional phenotypes: Proliferation, apoptosis/repair markers, ORR/PFS
Controls & Confounders
Arms: Signal only | Chromatin only | AKG only | Full triad
Confounders: Feedback activation (RTKs/MAPK), metabolic compensation, off-target HDAC effects
Monitoring: Phospho-proteomics, metabolomics
Reproducibility & Data Transparency
- Success thresholds: ≥2 independent loci re-expressed with concordant chromatin opening and demethylation
- Data deposition: GEO/PRIDE
- Blinded pathology reads
Prior Signals to Replicate/Extend
- Case-level documentation of multi-chromosome re-expression (ESR1/PGR) in refractory human disease using the triad
- Banner mechanism: “PI3K inhibition preserving mTOR-S6 repair + SIN3-HDAC relief + AKG-mediated de-repression”
Why This is Falsifiable
- Lack of chromatin accessibility (ATAC/ChIP)
- No shift in 5-mC/5-hmC despite AKG support
- Failure of receptor protein to reappear despite transcriptional signals
- Complete PI3K suppression reducing mTOR-linked repair and worsening DDR metrics
Note: This is not a clinical protocol. Investigational use requires proper approvals and safety oversight.
Conclusion
This framework outlines a mechanistically grounded, step-wise, and falsifiable strategy for multi-locus gene re-expression in vivo. By integrating signal modulation, chromatin remodeling, and metabolic/epigenetic support, researchers can systematically test hypotheses regarding transcriptional reactivation without permanent genome editing. Predefined readouts, rigorous controls, and transparent reporting ensure reproducibility, creating a robust translational path from bench to potential clinical impact.
References
- Engelman, J.A., Luo, J., Cantley, L.C. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–619.
- Kouzarides, T. Chromatin modifications and their function. Cell. 2007;128:693–705.
- Cedar, H., Bergman, Y. Programming of DNA methylation patterns. Annu Rev Biochem. 2012;81:97–117.
- Sabari, B.R., et al. Intracellular α-ketoglutarate links metabolism to epigenetic regulation. Nat Rev Mol Cell Biol. 2017;18:1–15.
- Phillips-Cremins, J.E., Corces, V.G. Chromatin insulators: linking genome organization to cellular function. Mol Cell. 2013;50:461–474.
- Loven, J., et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–334.
- Morris, J.R., et al. The role of mTOR in DNA damage response and repair. Nat Rev Mol Cell Biol. 2010;11:343–354.
- Airmid Biogenetics. Case reports on multi-locus receptor re-expression. ResearchGate; 2022.
